Co to jest zespół Ehlersa-Danlosa?
Zespół Ehlersa-Danlosa (Ehlers-Danlose Syndrom – EDS) to grupa chorób przebiegających z zaburzeniami metabolicznymi tkanki łącznej oraz defektami kolagenu. Z tkanki łącznej zbudowane są m.in. kości, ścięgna, więzadła i krążki międzykręgowe. Synteza nieprawidłowych form kolagenu prowadzi do nadmiernej ruchliwości stawów oraz wielu zaburzeń w obrębie skóry, naczyń krwionośnych i narządów wewnętrznych.
EDS ma podłoże genetyczne i jest wywołane mutacjami pojedynczych genów. Wyróżnia się wiele postaci zespołu Ehlersa-Danlosa, w tym postać: klasyczną, hipermobilną, naczyniową, sercowo-zastawkową, skórną czy kifoskoliotyczną. Każdy z typów EDS ma odmienny obraz kliniczny, w tym zróżnicowane objawy, przebieg i rokowania. W zależności od rodzaju, choroba jest dziedziczona w sposób autosomalny dominujący lub autosomalny recesywny. Postać klasyczna schorzenia występuje średnio u jednej osoby na 20000 przypadków, natomiast postać naczyniowa pojawia się z częstotliwością 1/100000. Nie zaobserwowano różnic między liczbą zachorowań wśród kobiet i mężczyzn.


Rolki

Objawy zespołu Ehlersa-Danlosa
Objawy zespołu Ehlersa-Danlosa zależą od typu schorzenia. Najczęstszymi postaciami choroby są: postać klasyczna, która dotyczy czterech na pięciu pacjentów z EDS oraz postać z nadmierną ruchliwością stawów (w dawnej klasyfikacji oznaczana jako typ III). U osób z zespołem Ehlersa-Danlosa obserwuje się hipermobilność stawów biodrowych, kolanowych, łokciowych i ramiennych. Chorzy mogą przyciągnąć kciuk do przedramienia, wyginać palce w stronę grzbietową oraz dotykać nos językiem.
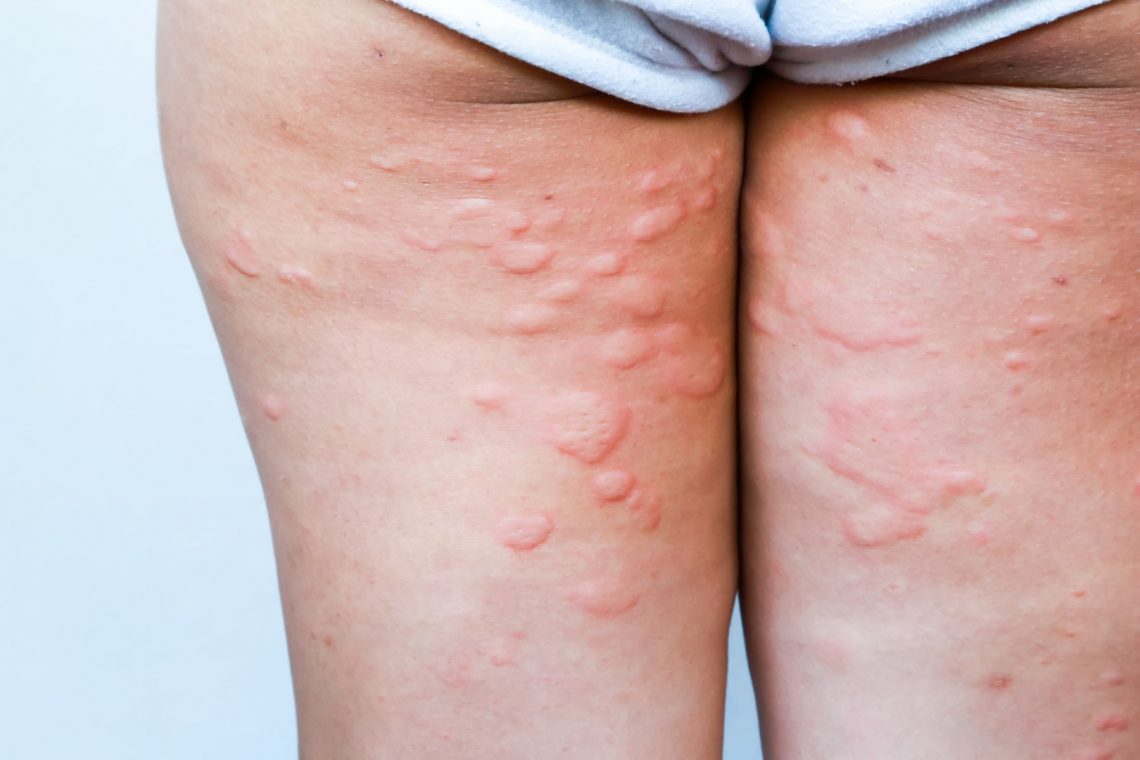
Ponadto występują przeprosty w stawach łokciowych i kolanowych, płaskostopie oraz charakterystyczny wygląd twarzy: jest ona szczupła i pociągła, nos ma szeroką nasadę, policzki są zapadnięte, a oczy wypukłe. Nadmierna ruchliwość stawów jest przyczyną częstych zwichnięć i skręceń, a także uszkodzeń skóry, w tym jej pękania, tworzenia się siniaków i wybroczyn. Ze względu na trudne gojenie się ran, często dochodzi do tworzenia się blizn i przebarwień.
Inne objawy zespołu Ehlersa Danlosa to:
- wady serca: niedomykalność zastawki dwudzielnej i aortalnej, hipotonia mięśniowa – w typie sercowo-zastawkowym,
- cienka, prześwitująca skóra, kruche naczynia krwionośne, żylaki występujące w młodym wieku, rozległe podbiegnięcia krwawe, zaniki dziąseł – w typie naczyniowym,
- skrzywienie kręgosłupa, rozciągliwość skóry, spadek masy mięśniowej, pęknięcia twardówki – w typie kifoskoliotycznym,
- podatność skóry na uszkodzenia, błękitne twardówki, nadmiar zwisającej skóry, częste występowanie przepuklin – w typie skórnym,
- marszczenie się skóry na twarzy, zapalenie przyzębia – w typie z objawami przedwczesnego starzenia się,
- nadmierna ruchliwość stawów, wrażliwość skóry na urazy, skrzywienie kręgosłupa, zanikowe blizny, zaokrąglenie twarzy, osteopenia – w typie z wiotkością stawów.

Zespół Ehlersa-Danlosa u dzieci
Zespół EDS występuje od urodzenia, ale pierwsze symptomy schorzenia często obserwuje się dopiero w okresie dojrzewania albo po doznanym urazie. Dzieci z hipermobilną postacią choroby są bardziej giętkie niż ich rówieśnicy, a niestabilność stawów doprowadza u nich do częstych zwichnięć i podwichnięć. Ponadto występuje wysoka podatność na urazy, tworzenie się siniaków i długo gojących się ran. Problemy z poruszaniem się i zmniejszona siła mięśniowa mogą utrudniać noszenie ciężkich plecaków. Należy dążyć do tego, aby dzieci z EDS prowadziły możliwie jak najbardziej „normalny” tryb życia i nie rezygnowały z aktywności fizycznej, o ile nie prowadzi ona do nadmiernego obciążenia stawów i wzrostu ryzyka kontuzji.
Diagnostyka zespołu Ehlersa-Danlosa
W diagnostyce zespołu Ehlersa-Danlosa konieczne jest zebranie dokładnego wywiadu rodzinnego oraz wykonanie badania przedmiotowego, w którym ocenia się m.in. wygląd i elastyczność skóry oraz ruchomość stawów wg kryteriów Beightona. Następnie zostają zlecone badania laboratoryjne pozwalające na wykrycie nieprawidłowych włókien kolagenowych. Rozpoznanie stawia się na podstawie objawów klinicznych, badań biochemicznych, badań genetycznych oraz diagnostyki różnicowej (różnicowanie EDS z zespołem Marfana, zespołem Menkesa oraz zespołem łagodnej nadruchliwości stawów).
Rodzaj dostępnych testów genetycznych zależy od wybranej placówki medycznej. Jedną z metod diagnostycznych jest metoda sekwencjonowania nowej generacji (NGS) umożliwiająca ocenę kilkudziesięciu genów. Materiałem badawczym jest ślina lub krew żylna. Cena badań genetycznych w kierunku zespołu Ehlersa-Danlosa wynosi średnio 2,500-3,600 złotych.

Leczenie i rokowania w chorobie Ehlersa-Danlosa
Ze względu na charakter choroby, leczenie przyczynowe zespołu Ehlersa-Danlosa nie jest możliwe. Terapia polega na leczeniu objawowym, a zastosowana farmakoterapia ma działanie przeciwzapalne i przeciwbólowe. Chorym zaleca się dietę zawierającą optymalną ilość witaminy C, która jest konieczna do wytwarzania kolagenu. Ponadto podawany jest kwas hialuronowy oraz witamina D. Pacjent powinien zostać objęty holistyczną opieką wielu specjalistów, w tym kardiologa, reumatologa i ortopedy.
Celem leczenia EDS jest ochrona narządu ruchu, którą można uzyskać poprzez zajęcia fizjoterapeutyczne, fizykoterapię oraz stosowanie sprzętu i akcesoriów ortopedycznych. Wkładki ortopedyczne pomagają w prawidłowym ustawieniu stopy oraz zmniejszają napięcie mięśniowe. Niektórzy chorzy korzystają również ze stabilizatorów. Dzięki rehabilitacji możliwe jest zwiększenie siły mięśniowej oraz stabilizacja stawów i kręgosłupa.
Przebieg i rokowania w chorobie EDS są trudne do przewidzenia, ponieważ zależą od typu schorzenia, manifestacji objawów klinicznych oraz występujących powikłań. Uznaje się, że chorzy z klasyczną postacią zespołu Ehlersa-Danlosa przeżywają przeciętną długość życia, jednak ich jakość życia jest pogorszona. Najgorsze rokowania występują w typie naczyniowym ze względu na wysokie ryzyko śmiertelnych powikłań.
Bibliografia:
- P. Gajewski, A. Szczeklik, Interna Szczeklika, MP, 2017.
- F. Malfait i wsp., The 2017 International Classification of the Ehlers–Danlos Syndromes,American Journal of Medical Genetics Part C (Seminars in Medical Genetics) 175C:8–26, 2017.
- I. Dereń-Wagemann i wsp., Ehlers-Danlos Syndrome, Adv Clin Exp Med , 19, 4, 537–542, 2010.